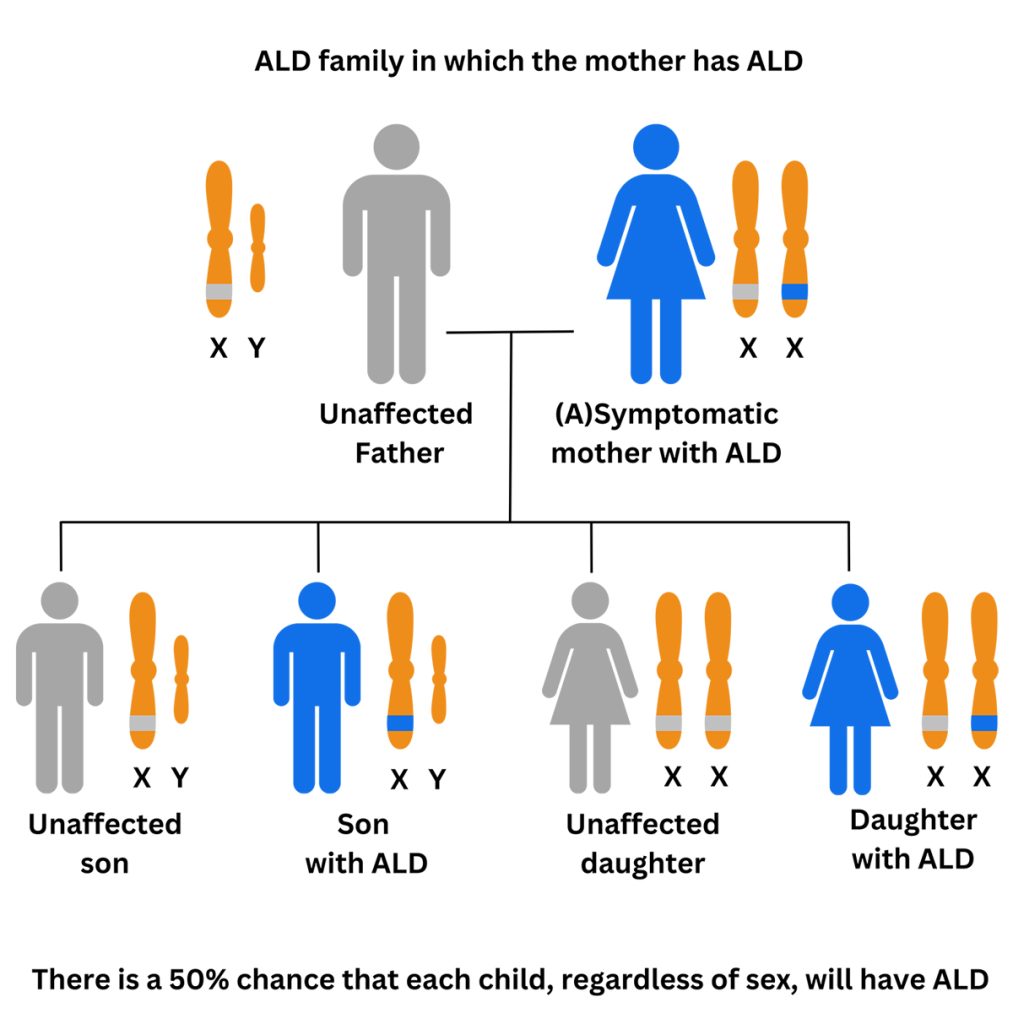
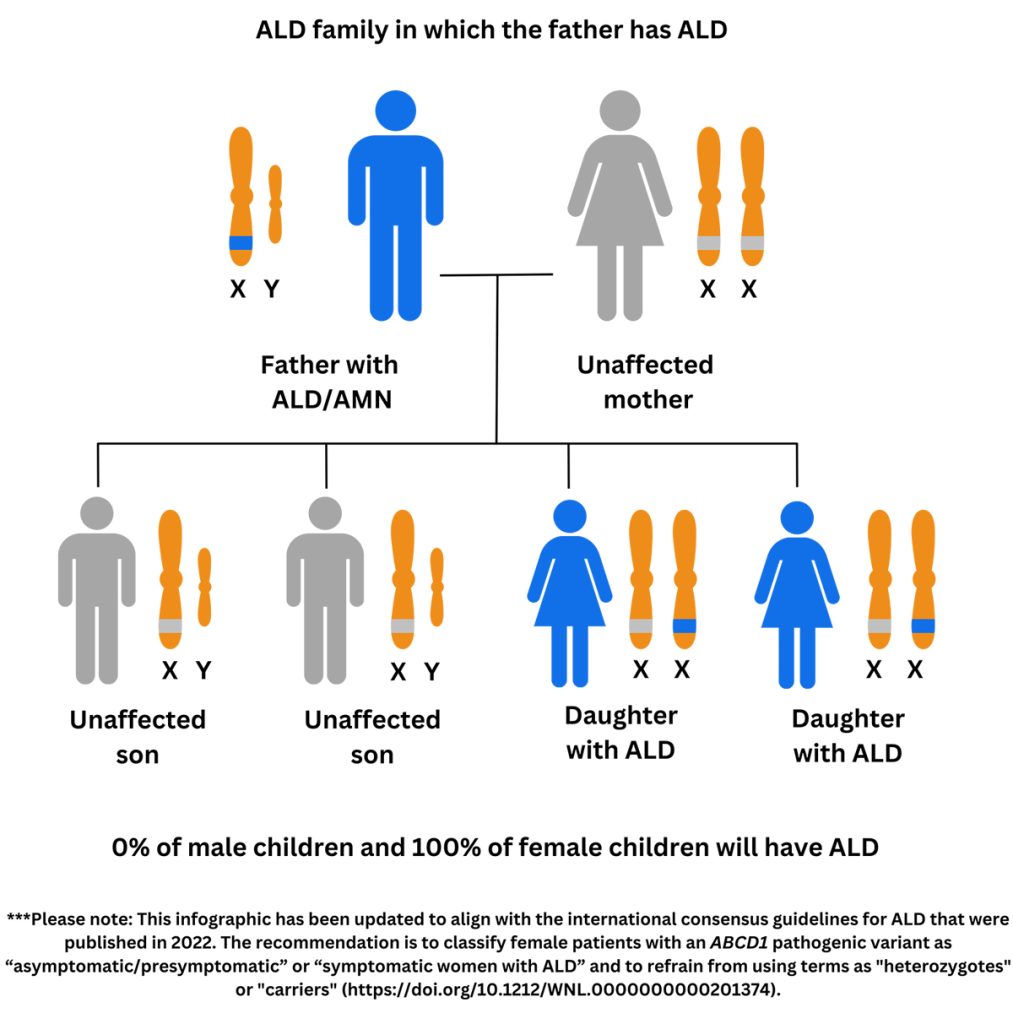
Genetic testing, which looks for a mutation (also known as a variant) in the ABCD1 gene, is considered the gold standard for ALD diagnosis. Talk to your doctor or genetic counselor to start the genetic testing process and for help understanding the results. Genetic testing will identify the specific variant in your family (i.e. how the spelling of the ABCD1 gene is changed in your family). If a variant is found in the ABCD1 gene that is known to cause disease or is very likely to cause disease, it will be considered “pathogenic” or “likely pathogenic”, which will confirm the diagnosis of ALD.
Sometimes, a variant will be detected in the ABCD1 gene that has not been seen before in symptomatic patients with ALD, and there is not enough evidence to yet know if this variant will disrupt the function of the gene and be “pathogenic”, or if it will result in no functional consequence to the gene and be a “benign” variant. These variants with inadequate information will be called “Variants of Uncertain Significance (VUS)”, meaning it is unclear from the genetic information alone whether they will cause ALD. In the case of VUSes, it is particularly important to consider whether the patient has any symptoms and whether their biochemical markers are elevated (see below) to establish an ALD diagnosis. Due to the serious consequences of ALD manifestations, particularly adrenal insufficiency and cerebral ALD in boys, it is important that children with a VUS in the ABCD1 gene still undergo routine monitoring for ALD.
Traditional biochemical testing for ALD consists of fasting very long-chain fatty acid (VLCFA) testing. VLCFA testing measures the levels of VLCFAs in the plasma, and is very accurate for males with ALD and AMN, who have elevated levels of VLCFAs. This test can also be conducted on women, however 15% of women with ALD will have normal VLCFA levels, resulting in a “false negative” result.
More recently, an updated biochemical biomarker has become available for testing, known as C26:0-lysophosphatidylcholine (C26:0-lysoPC), which is extremely accurate in both men and women with ALD. In contrast to VLCFA testing, C26:0-lysoPC testing will detect more than 99% of women with ALD, and is thus considered more accurate, especially for women.
Newborn screening is a screening test conducted with dried blood spots taken from a baby just after birth. In many states, ALD is included on the newborn screening panel, and babies are screened for ALD using a modified VLCFA test. If a baby screens positive for ALD, they will have confirmatory diagnostic testing before being officially diagnosed with ALD.